

Did Pfizer Perform Adequate Safety Testing for its Covid-19 mRNA Vaccine in Preclinical Studies?
Evidence of Scientific and Regulatory Fraud
By Sasha Latypova
April 20, 2022
The rushed “warp speed” development and approval of completely novel Covid-19 mRNA and DNA vaccines pushed on millions of people has resulted today in millions of reported injuries and thousands of deaths according to public health databases such as VAERS (US), Eduravigilance (EU), Yellow Card (UK) and others. This article reviews some of the publicly available documents on Pfizer’s non-clinical development program and points to its deficiencies, omissions and gaps that were clearly visible, yet never questioned by the regulators or other health authorities. The cursory nature of the entire preclinical program can be briefly summarized as “we did not find any safety signals because we did not look for them”. The omissions of standard safety studies and glaring scientific dishonesty in the studies that were performed are so obvious that they cannot be attributed to the incompetence of the manufacturers and regulators. Rather, the question of wilful negligence should be raised.
The focus of my review was the scope and adequacy of the program of non-clinical assessment for a novel gene therapy vaccine with a brief discussion of relevant regulatory frameworks. I did not dive deeply into the review of the results of specific studies. My goal is to illustrate the complete breakdown of the previously known to be rigorous ethical drug development process, as well as the shocking negligence on the part of the regulatory agencies that are supposed to keep the pharmaceutical manufacturers honest. It turns out that both were highly dishonest and pushed an entirely novel technology and product on millions of people without a single well designed safety assessment.
In summary, I have identified the following:
- Finding 1: Pfizer’s program did not include a comprehensive end-to-end test of all components of the final approved product. The studies included in the approval package were for a variety of versions of the product with no comparability assessments, thus no comprehensive assessment of product safety can be made.
- Finding 2: The toxicity of the Covid 19 vaccine’s mRNA active ingredient was never studied!
- Finding 3: Pfizer claimed absence of potential for enhanced covid illness based on the animal study where no covid illness was observed.
- Finding 4: CDC, FDA and Pfizer lied about “vaccine staying in the injection site”.
- Finding 5: Pfizer skipped major categories of safety testing altogether.
- Finding 6: Pfizer used dishonest and self-serving interpretation of regulatory guidelines to avoid routine safety testing.
- Finding 7: Both FDA and Pfizer knew about major toxicities associated with gene therapy class of medicines, and therefore cannot claim lack of anticipatory knowledge of these risks. This points to intentional fraud and collusion between Pfizer and the regulators to push this untested dangerous product on the market.
Background:
Pre-clinical studies are the initial phase of Pharmaceutical R&D process, where a drug/biological product is tested on cell lines, small animals (e.g., mice and rats), and larger animals (e.g., monkeys or other species). The aim of preclinical studies is to demonstrate Proof-of-Concept and characterize safety and efficacy to a “sufficient” level. For traditional vaccines, non-clinical phase is the only phase of development where safety and toxicity are formally assessed. The phases of drug development have been defined for purposes of de-risking the new medicines, reducing potential for harm to human subjects, and ensuring an improved understanding of its risk/benefit with every step.
The US Food and Drug Administration (FDA) guidelines for different phases of development are described in FDA Guidance for Industry publications1 and are harmonized globally via International Conference on Harmonization (ICH). Separate sets of guidelines are issued for conventional drugs (small molecules) and biological products, including vaccines. However, for more complex platforms, such as drug-device or drug-biologics combinations, both types of guidelines are applicable depending on the finished product composition. Since 2013, the FDA has been publishing guidelines that are directed at gene therapy platforms specifically. The most recent FDA Guidance on early phase testing of gene therapy products was published in 2015: “Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products”.2 Relevant aspects of this guidance and FDA position on this class of drugs is discussed at the end of this paper (Finding 7) and demonstrate what the FDA and manufacturers knew about the risks associated with the class prior to 2020.
The exact scope of each new medicine’s development program is negotiated in a series of meetings between the manufacturer and the FDA. In general, the more novel the new drug or biologics entity, the more stringent and extensive the testing that is required due to lack of well characterized risks and safety data from prior experience with the class or type of product. Numerous unknowns and thus potential for harms in patients need to be assessed, risks, if identified must be characterized so that a well-informed risk/benefit assessment can be performed. A drug or a biological product is deemed dangerous until proven safe. Claiming something is safe purely on theoretical grounds or because “all vaccines are safe” is not scientifically or ethically acceptable. In addition, it should be noted the FDA has not historically permitted the testing of different versions of candidate product under the same IND application.
A well designed non-clinical testing strategy will contain characterization of the product and its components in the following general categories of research (not complete list of topics, description of each is provided further in the article):
- Pharmacokinetics/Pharmacodynamics (drug distribution and the processing of it by the body)
- Pharmacology (mechanism of action), including primary and secondary (off target) effects
- Safety Pharmacology and Toxicology, including characterization of risks for major organ classes (cardiovascular, CNS, hepatic, renal, blood and other organ systems selected based on known or predicted effects of the product class or its components)
- Genotoxicity (proclivity to cause damage to genetic material)
- Carcinogenicity (proclivity to cause cancer)
- Reproductive Toxicology (before the product can be administered to people of reproductive potential)
- Other categories of studies designed to characterize the risk based on previously identified safety signals
Further, while global agencies such as World Health Organization may provide technical or scientific opinion via published recommendations, in the United States the sole authority to regulate drugs/biologics development and approve new products is vested with the FDA.
Pfizer’s Non-Clinical Program for Covid-19 mRNA Vaccine:
Recently, some of the documents used by FDA to approve Pfizer’s mRNA platform based Covid-19 Vaccine have been obtained via Freedom or Information lawsuits, overcoming FDA and Pfizer’s motions to keep this information secret for 75 years. One package of these documents, titled “BNT162b2 Module 2.4. Nonclinical Overview” (466 pages) was obtained by Judicial Watch from HHS.3
Finding 1: Pfizer’s program did not include a comprehensive end-to-end test of all components of the final approved product. The studies included in the approval package were for a variety of versions of the product with no comparability assessments, thus no comprehensive assessment of product safety can be made.
On p. 6 Pfizer documents state that (emphasis added):
BNT162b2 (BioNTech code number BNT162, Pfizer code number PF-07302048) is an investigational vaccine intended to prevent COVID-19, which is caused by SARS-CoV-2. BNT162b2 is a nucleoside modified mRNA (modRNA) expressing full-length S with two proline mutations (P2) to lock the transmembrane protein in an antigenically optimal prefusion conformation (Pallesen et al, 2017; Wrapp et al, 2020). The vaccine is formulated in lipid nanoparticles (LNPs). The LNP is composed of 4 lipids: ALC-0315, ALC-0159, DSPC, and cholesterol. Other excipients in the formulation include sucrose, NaCl, KCl, Na2HPO4, and KH2PO4. The dose selected for BNT162b2, with efficacy demonstrated in Phase 2/3 clinical evaluation and intended for commercial use, is 30 µg administered IM as two doses given 21 days apart.
It is clear from the description of the product that this completely novel platform consists of new proprietary biological, genetic, and chemical components comprising a “payload + delivery vehicle” structure. In situations where complex products containing combinations of drugs and biologics, or biologics and novel delivery vehicles (such as Pfizer product), the manufacturer is required to assess safety of all components separately and in the final assembled version which is intended for human phases of development.4
Pfizer’s document further explains that several versions of the product were tested preclinically, and one selected to be studied in clinical trials, and that specific vaccine candidate BNT162b2 V9 is therefore the subject of the Biologics License Application (BLA), i.e. the product that was ultimately fully licensed for market by the FDA in 2021:

The highlighted statement above is not true. Review of clinical studies released by FOIA uncovered that at least 4 different categories of active ingredient were included in the single Investigational New Drug application by Pfizer IND#19736:
- SARS-CoV-2 Spike Protein;
- BNT162a1 (uRNA; variant RBL063.3);
- BNT162b1 (modRNA; variant RBP020.3);
- BNT162b2 (modRNA; variant RBP020.2);
- BNT162c2 (saRNA; variant RBS004.2)
Each category can be delivered by the Lipid Nanoparticles (ALC-0315, ALC0159, DSPC and Cholesterol).5
Furthermore, it is evident from the Phase 1/2/3 clinical trial protocol, amendments and the Investigator’s Brochure that in various parts of the study and different geographies, Pfizer used several versions of the mRNA active ingredient6, and new versions not mentioned in the IND above were added (e.g. South Africa specific variant in 2021).
Note that while multiple versions of a product in early stages of development are often inevitable, for the purpose of product approval, each chemical or biological entity is deemed legally distinct, and therefore studies conducted with versions of the product that are not the exact specification of the final approved product may provide supporting information but should not be deemed definitive tests for the safety or efficacy claims. In September 2021 the FDA has issued a draft guidance: “Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial” stating that each version of product requires a separate IND, however in a footnote they stated that this does not apply to “vaccines intended to prevent infectious diseases”. No explanation is given on why this exception is made and no scientific or legal basis exists for this exception, other than the FDA has arbitrarily allowed this dramatic unprecedented deviation from the regulatory standard and now needs to cover their tracks. In fact, one can make an argument that this regulatory “exception” does not even apply to Pfizer’s covid 19 “vaccine” since the product does not prevent infection or transmission of the disease.
Pfizer states that the primary pharmacology, distribution, metabolism, safety, and immunogenicity of BNT162b2 were studied in-vitro and in-vivo in mice, rats and rhesus monkeys as well as several cell-culture assay experiments. There was a total of 18 studies included in the Non-Clinical package, of which 7 were for the V9 candidate (including 1 non-GLP7 study which should not be acceptable for labelling), and 6 studies were for only two of the four lipid excipients (ALC-0315 and ALC-0159). The other lipids included in the Lipid Nanoparticle Platform (LNP) – DSPC and cholesterol were not studied. Of the 6 studies of the lipid excipients, 4 were for lipid formulations “comparable to LNP in BNT162b2” and 2 of the studies were non-GLP. It is not explained anywhere in the document how these formulations differed from the final formulation of the LNP included into the approved product, and how their comparability was determined, other than Pfizer’s assertion that they were comparable.
Therefore, only 9 of the 18 studies in this package are directly related to the licensed product and to only some of the components of the final product.
Finding 2: The toxicity of the vaccine’s mRNA active ingredient was never studied!
In the studies designed to test whether the vaccine remains near the injection site or travels throughout the body, Pfizer did not use the test article representative of the commercial vaccine (BNT162b2 – mRNA coding for full-length spike protein with two proline mutations P2) but instead, studied biodistribution by administering “modRNA encoding luciferase formulated in LNP comparable to BNT162b2 with trace amounts of [3H]-CHE as nondiffusible label” to mice and rats – a “surrogate” mRNA producing the luciferase protein. This surrogate is clearly not coding for spike protein, and therefore no conclusions can be made whether the theoretical promise of no circulation of spike protein in the blood stream is indeed true. Additionally, the LNP delivery formulation used is not the same, but “close enough” to the final vaccine product. The claims unsupported by any data in the non-study of only half of the product formulation are breathtakingly scientifically dishonest:
“Distribution to the liver is likely mediated by LNPs entering the blood stream. The luciferase expression at the injection sites dropped to background levels after 9 days. The repeat-dose toxicity study in rats showed no evidence of liver injury (Section 2.4.4.3). The biodistribution of the antigen encoded by the RNA component of BNT162b2 is expected to be dependent on the LNP distribution and the results presented should be representative for the vaccine RNA platform, as the LNP-formulated luciferase-encoding modRNA had the same lipid composition.”
Let’s reiterate: Pfizer has evaluated an unrelated product surrogate and made the claim that the toxicity and safety results are representative of its mRNA active ingredient encoding spike protein! More shocking still, the FDA did not find this objectionable.
No rationale for not testing the mRNA coding for spike protein is provided. It would have been possible to assess the expression (and subsequent effects or lack thereof) of the spike protein in various tissues of interest. After all, the recent histopathological studies based on autopsies of post-vaccine deaths8 provide ample evidence that the expression of spike protein with subsequent organ damage was possible to detect and study with standard techniques.
Finding 3: Pfizer claimed absence of potential for enhanced covid illness based on the animal study where no covid illness was observed.
Disease enhancement by mRNA or DNA based vaccination is a known risk factor that has been identified in numerous prior animal studies for gene therapy class. Pfizer and FDA were clearly aware of this risk. However, in the non-clinical package, the manufacturer claimed “no evidence of vaccine-elicited disease enhancement” in the single animal study of 6 rhesus monkeys where no clinical symptoms of covid illness were elicited in the first place, in either inoculated or control animals after virus challenge. The study of immunogenicity (VR-VTR-10671) was conducted in 6 rhesus monkeys inoculated with 2 injections 21 days apart, and a control group of 3 monkeys. While inoculated monkeys produced the measurable antibody response, and upon viral challenge, produced much lower levels of viral RNA in the lungs vs the control group, none of the monkeys got clinically ill. Pfizer states that (emphasis added): “None of the challenged animals showed clinical signs of significant illness, indicating that the 2-4 years old male rhesus challenge model is primarily an infection model for SARS-CoV-2, not a COVID-19 disease model”. It seems that for Pfizer (and FDA), studying a non-disease model to make claims about lack of the enhanced disease is acceptable.
Finding 4: CDC, FDA and Pfizer lied about “vaccine staying in the injection site”.
The biodistribution study in rats clearly shows that the payload (whatever it happens to be – luciferase surrogate or never-tested mRNA encoding for spike protein) is getting into the bloodstream and is being distributed all over the body: there are major accumulations in adrenal glands, liver, spleen, ovaries, and other organs such as lymph nodes and bone marrow as the table below illustrates. In fact, one of the studies in rats included in the package anticipates the product reaching the blood stream directly and includes the IV route of administration. This study is also based on the surrogate mRNA and not the spike protein encoding version.
Since the focus of this article is the scope of Pfizer’s non-clinical program, and not in-depth review of these studies, I refer the readers to an excellent analysis performed by scientific experts in this field.9
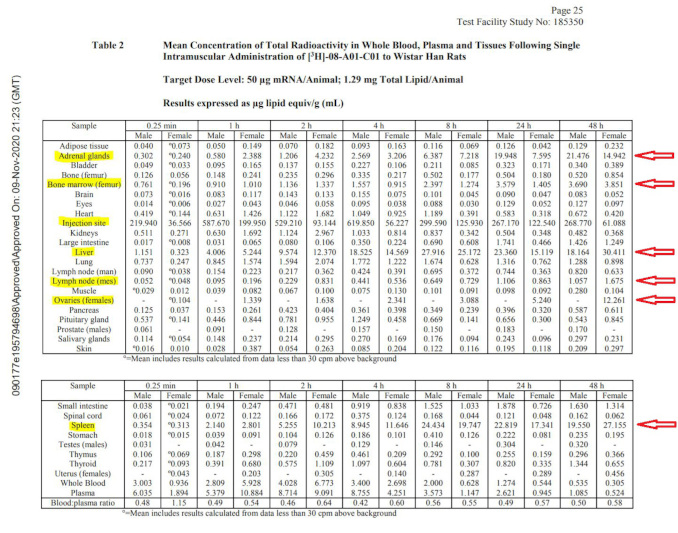
Notably, this study is incomplete: it does not fully characterize the biodistribution of the LNPs carrying their payload. The study was stopped while the concentrations in the liver, spleen and ovaries of the females were still increasing, and therefore it is not possible to say what maximum concentrations in various organs were observed. No follow-on studies elucidating the complete time course of distribution, time to maximum concentration, maximum concentrations observed, and time to clearance were performed or planned. No estimates of the therapeutic safety margins were provided.
The overall nonclinical testing program appears woefully incomplete. This fact was clearly noted in the European Medicines Agency (EMA) summary document10 of the “Comirnaty” BNT162b2 vaccine. The reviewers share an explicit admission that “No traditional pharmacokinetic or biodistribution studies have been performed with the vaccine candidate BNT162b2.” Additionally, the EMA document states “Biodistribution: Several literature reports indicate that LNP-formulated RNAs can distribute rather nonspecifically to several organs such as spleen, heart, kidney, lung, and brain. In line with this, results from the newly transmitted study 185350, indicate a broader biodistribution pattern.”
Although not performed to industry GLP standards, these results seem to indicate that lipid nanoparticles with mRNA which codes for the spike protein, reach the bloodstream, circulate throughout the body, and then collect in a variety of organs and tissues. If this results in spike protein expressed in those organs, they will both stimulate immunity and cause those same cells to be attacked by the immune system. The resulting “vaccine reactogenicity” could resemble clinical symptoms seen with autoimmune syndromes of various severity, in some cases severe enough to cause death or permanent disability. With the rollout of the vaccines globally, these exact types of adverse events have been reported in thousands in the vaccine adverse event reporting systems, yet no public health agency has yet made a connection between this preclinically documented mechanism and the alarming current health outcomes data.
Finding 5: Pfizer skipped major categories of safety testing altogether.
Even more elucidating is what Pfizer chose NOT to study, i.e., the entire pharmacology sections related to safety and risk characterization. Specifically, the Non-Clinical document package states that:
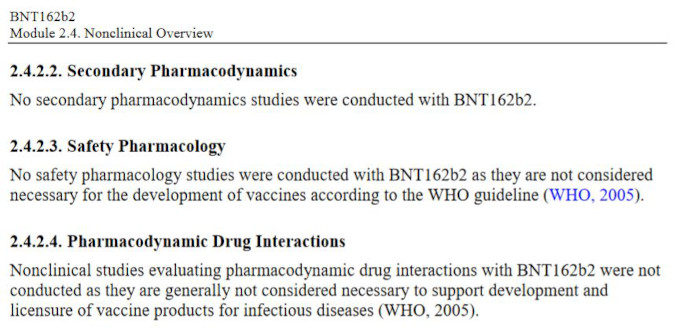

Let’s review what safety studies Pfizer decided to omit entirely:
What is Safety Pharmacology?
The aim of Safety Pharmacology is to characterize the pharmacodynamic/pharmacokinetic (PK/PD) relationship of a drug’s adverse effects. Pharmacodynamics aim to describe how the drug acts on the body while Pharmacokineticshow the body acts on the drug. The structure of a Safety Pharmacology ‘core battery’ studies is to determine the potential undesirable effects of a drug on the central nervous, cardiovascular, and respiratory systems, as well as to implement supplementary tests to evaluate other organ systems (liver, kidney, blood, etc) if there is a potential for risks or damage to these systems.
What are Secondary Pharmacodynamics Studies?
Assessments of new drugs for pharmacological activities other than the desired therapeutic target are called Secondary Pharmacology. For an entirely novel class of biological compound with a completely novel and undisclosed excipients it is unacceptable to omit evaluation of secondary pharmacodynamics of the whole product and/or its components. In the case of BNT162b2, it can be argued that neither primary nor secondary pharmacology studies were performed. Primary pharmacology of the mRNA encoding for spike protein was never assessed, and the secondary studies were simply dismissed as unnecessary by Pfizer.
What are Drug Interaction Studies?
Drug interactions studies are designed to assess potential harmful effects from interactions of the novel pharmaceutical product with existing medicines which a patient may be taking. This considers additional burden on major metabolizing organs, such as liver.
What are Genotoxicity and Carcinogenicity Studies?
Studies designed to evaluate the risk of possible damage to DNA cellular processes, and to evaluate the risk of promoting cellular damage and cancer formation. DNA/RNA based novel technologies have an obvious potential to cause mutagenic effects, and the cationic lipids are known to be cytotoxic, i.e., have the potential to cause cancer11. The product was never tested to determine if it causes cancer – neither in animals nor in humans. The next logical question is what rationale was used to waive these entire categories of pharmacological safety testing?
The next logical question is what rationale was used to waive these entire categories of pharmacological safety testing?
Finding 6: Pfizer used dishonest and self-serving interpretation of regulatory guidelines to avoid routine safety testing.
Numerous mechanisms injury on major organ systems12 , 13 , 14 , 15 as well as previous failed animal experiments with mRNA platform were documented16 , 17 yet Pfizer claimed to the FDA that Safety Pharmacology, Secondary Pharmacodynamic, Genotoxicity or Carcinogenicity studies were not necessary for their product, and as justification of this claim, Pfizer cited World Health Organization’s Guidelines for Vaccine Development from 2005.
Pfizer’s product was only arbitrarily reclassified as a vaccine in 2020. Prior to that it was categorized as a gene therapy, so back in 2005 when the WHO guidelines were written, it would not have been regarded as a vaccine.
Furthermore, 2005 recommendations from WHO did not anticipate gene therapy platforms. Additionally, it is the responsibility of the FDA and other regulatory bodies worldwide to regulate the authorization and licensing of medical products. WHO does not have this authority as it is only an advisory and coordination non-governmental body.
What Do WHO Recommendations from 2005 Really State?
WHO Guidelines18 on nonclinical evaluation of vaccines state that PK studies are not normally needed but should be considered on a case-by-case basis (para 4.2.6), and toxicity studies should be performed where new excipients (and preservatives) are used for which no toxicological data exist (para 5.2). Para 4.2.4 states: “Safety pharmacology. The purpose of safety pharmacology is to investigate the effects of the candidate vaccine on vital functions. If data from nonclinical and/or human clinical studies suggest that the vaccine (e.g. one based on specific toxoids) may affect physiological functions (e.g. central nervous system, respiratory, cardiovascular and renal functions) other than those of the immune system, safety pharmacology studies should be incorporated into the toxicity assessment. Useful information on this topic can be found in the Note for Guidance on safety pharmacology studies for human pharmaceuticals”.
Finding 7: Both FDA and Pfizer knew about major toxicities associated with gene therapy class of medicines, and therefore cannot claim lack of anticipatory knowledge of these risks. This points to intentional fraud and collusion between Pfizer and the regulators to push this untested dangerous product on the market.
Several FDA Guidance documents exist for studying investigational cellular and gene therapy products, including therapeutic vaccines, specifically:
- Preclinical Assessment of Investigational Cellular and Gene Therapy Products (2013),
- Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products (2015)
- Development and Licensure of Vaccines to Prevent COVID-19 (June 2020)
- Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial (Draft, September 2021)
- Human Gene Therapy Products Incorporating Human Genome Editing (Draft, March 2022)
These guidance documents contain regulatory thinking that clearly anticipates many risks with this class of product. Specifically, the guidance on non-clinical studies from 2013 states that:
“Use of in vitro studies is strongly encouraged for identification of potential safety issues and MOA [mechanism of action] of an investigational CGT product. However, this testing alone is not sufficient to reliably anticipate the outcome of physiological and functional integration of the product following in vivo administration. Accordingly, the preclinical testing program should incorporate a stepwise, multifactorial approach to achieve an understanding of the biological plausibility for use of the investigational CGT product in the intended patient population. For in vivo preclinical testing, the use of animal models of disease/injury is encouraged, as such studies allow for the characterization of resulting morphological changes in conjunction with observable functional/behavioural changes.”
FDA guidance on early-phase clinical trials program from 2015 is extensive and warns of severe known risks from prior experience with gene therapies:
- Multi-organ failure and death
- Potential for tumors/cancer development
- Late onset T-cell leukemia
- Potential for prolonged uncontrollable activity after single administration
- Immunogenicity as a risk (autoimmunity)
- Uncontrolled expression of genes
- Migration of product to undesired organ systems
- Possibility of shedding: excretion/secretion of viral particles that could be transmitted to other individuals
- Studies in healthy volunteers are not generally advised due to potential severe toxicities
The guidance also states that the risks associated with the gene therapy class may be entirely novel and cannot be derived from prior history of other drug classes. In other words, this class is uniquely risky and requires an extensive and rigorous safety testing program. It should be noted that prior to 2020, all gene therapy derived products were being developed for extremely severe, often terminal illnesses like terminal cancer and Huntington’s disease. They could not be even tested in healthy people, much less prescribed to every human on the planet as a prophylactic treatment, and much less forced on every human being regardless of consent.
Pfizer’s Non-Clinical document states that it considered FDA’s “Development and Licensure of Vaccines to Prevent COVID-19 Guidance for Industry” (2020)19. It’s not clear that any consideration of this document in fact took place since none of the recommendations from the FDA guidance were utilized in Pfizer’s nonclinical assessments. It’s worth asking why the WHO recommendations from 2005 and not FDA industry guidance document from 2020 was used as the basis for design of the non-clinical testing program? Specifically, the FDA guidance clearly states that:
“For a COVID-19 vaccine candidate consisting of a novel product type and for which no prior nonclinical and clinical data are available, nonclinical safety studies will be required prior to proceeding to FIH clinical trials 21 CFR 312.23(a)(8).”
FDA specifically expresses concern about the vaccine-associated enhanced respiratory disease and the need to characterize/exclude the risk with the novel vaccine product:
“Data from studies in animal models administered certain vaccine constructs against other coronaviruses (SARS-CoV and MERS-CoV) have raised concerns of a theoretical risk for COVID-19 vaccine-associated enhanced respiratory disease (ERD). In these studies, animal models were administered vaccine constructs against other coronaviruses and subsequently challenged with the respective wildtype virus. These studies have shown evidence of immunopathologic lung reactions characteristic of a Th-2 type hypersensitivity similar to ERD described in infants and animals that were administered formalin-inactivated respiratory syncytial virus (RSV) vaccine and that were subsequently challenged with RSV virus due to natural exposure or in the laboratory, respectively (Refs. 4-9). Vaccine candidates should be assessed in light of these studies…”
Given this knowledge of potential for disease enhancement, it is even more puzzling that Pfizer chose to disregard these guidelines, and FDA chose to ignore this disregard.
One paragraph from the guidance document describing when nonclinical safety studies might be waived caught my attention:
“In some cases, it may not be necessary to perform nonclinical safety studies prior to FIH clinical trials because adequate information to characterize product safety may be available from other sources. For example, if the COVID19 vaccine candidate is made using a platform technology utilized to manufacture a licensed vaccine or other previously studied investigational vaccines and is sufficiently characterized, it may be possible to use toxicology data (e.g., data from repeat dose toxicity studies, biodistribution studies) and clinical data accrued with other products using the same platform to support FIH clinical trials for that COVID-19 vaccine candidate. Vaccine manufacturers should summarize the findings and provide a rationale if considering using these data in lieu of performing nonclinical safety studies.”
It made my antennae go up. I am possibly jaded by the experience of government health authorities continuously lying, misrepresenting data, suppressing dissenting opinion and open scientific debate in the past few years. Nevertheless, I am questioning whether this paragraph is inserted to provide Pfizer and Moderna the future “out” of safety testing claiming their products are derivatives of “approved safe products” while erecting a regulatory barrier to other manufacturers who would wish to design a different Covid-19 vaccine?
Pfizer’s dishonest interpretation of guidelines and cherry-picking of the applicable regulations resulted in brazen disregard for all routine safety assessments.
It is unacceptable for a pharmaceutical manufacturer to not study their product for potential to harm major organ systems, not acceptable to substitute the product with a surrogate or a different version, claim theoretical comparability, and then assert that there are no risks to major human organ systems. Absence of evidence is not evidence of absence!
The mandate of the FDA as the industry regulator requires the agency to question and check such reckless disregard for safety testing. An honest regulator would have questioned the assertion by the manufacturer that major categories of safety studies were not applicable to their product. There is no question of incompetence. The agency is staffed with qualified and experienced pharmacology and toxicology professionals. At this point, with millions of adverse event reports accumulating rapidly in every public health database, neither the FDA, NIH, CDC, Pfizer nor other manufacturers can claim ignorance of these issues. The question of fraud and willful negligence by both the manufacturers and the regulators must be raised.
About the author:
Sasha Latypova is a Ukrainian-born entrepreneur living in the United States. Most of her professional career was in the pharma/healthcare industry with specific focus on development, validation, regulatory acceptance and commercialization of new clinical technologies and biomarkers.
Sasha Latypova’s channel on Bitchute with more presentations.

Footnotes:
1 https://www.fda.gov/vaccines-blood-biologics/guidance-compliance-regulatory-information-biologics
4 FDA Guidance for Industry: Principles of Premarket Pathways for Combination Products, January 2022
5 FDA Grant Fast Track Designation Letter, July 7, 2020.
7 GLP = Good Laboratory Practice for Nonclinical Laboratory Studies, Code of US Federal Regulations (21 CFR Part 58).
9 The Pfizer mRNA Vaccine: Pharmacokinetics and Toxicity
12 https://ijvtpr.com/index.php/IJVTPR/article/view/23/51
13 https://www.biorxiv.org/content/10.1101/2020.10.12.335083v1
14 https://www.nature.com/articles/nrmicro2090
15 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7547916/
16 https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0035421